Extending high-accuracy quantum chemistry calculations with HPC
This article originally appeared in R&D Magazine.
Researchers at the Argonne National Laboratory recently performed the largest, and most accurate calculation of a metal organic framework system that was ever done. What made this possible was the Theta supercomputer at the Argonne Leadership Computing Facility (ALCF) with the latest generation processors that incorporated extra memory on the chip to aid in memory intensive calculations.
The Argonne team is conducting critical quantum chemistry research relating to energy and semiconductors using the metallorganic framework. Their computer simulations can provide models for materials which are highly complex that might be used for applications such as energy generation, storage, and semiconductors.
Researchers seek to understand in depth chemical systems and chemical reactions in experiments using quantum chemistry. Theoretical quantum chemistry, often called computational chemistry, uses computers to calculate and predict the energy of atoms and molecules to understand the behavior and structure of materials and molecules. However, the accuracy required to do such calculations goes beyond what is achievable with classical physics/chemistry principles. Therefore, simulations must be carried out within the quantum mechanics framework. These types of simulations require high accuracy methods such as Quantum Monte Carlo implemented in highly efficient software such as QMCPACK and large scale HPC machines.
Argonne’s Breakthrough in Metallorganic Framework Calculation Research
The Argonne research team is performing innovative quantum chemistry research through the DOE-Office of Science – Basic Energy Research grant. Research simulations run at ALCF which is a Department of Energy (DOE) Office of Science User Facility, located at Argonne National Laboratory. Since 2006, the ALCF has operated state-of-the-art computing systems that are 10-100 times more powerful than systems typically available for open scientific research. In May 2016, ALCF was named as an Intel Parallel Computing Center (Intel PCC).
“Using optimized QMCPACK code running on ALCF’s Cray X40 supercomputer, Theta, our team was able to perform the largest highest accuracy calculation on a metal organic framework system, which is something that was never possible in research before,” Argonne scientist Anouar Benali and co-Principal Investigator of the Intel PCC at Argonne stated. “Our quantum chemistry metallorganic framework calculation requires 2 – 3 million core hours of computer processing. This would take between 50 – 100 years on a laptop. But we completed the computation in twelve hours using Theta running on its 4000 nodes.”
Optimizing Quantum Chemistry QMCPACK Code for Release to other Scientists
Quantum Monte Carlo techniques provide some of the most accurate solutions to quantum mechanical problems, allowing theoretical predictions for many problems at the forefront of research—from materials science to complex biological systems. One of the most popular quantum chemistry codes is the Quantum Monte Carlo (QMCPACK) open source code developed by the University of Illinois - Champagne and a consortium of DOE national laboratories. QMCPACK is a high-performance simulation code for solid-state physics and chemistry. Argonne’s scientific computing team works with Intel to optimize QMCPACK to take full advantage of massively parallel computer systems based on the Intel Many Integrated Core Architecture technology. The team used Intel VTune, Intel compilers and OpenMIC in optimizing and parallelizing the QMCPACK code. They have released these optimizations which are available in the latest version of the QMCPACK code.
Metallorganic Framework Quantum Chemistry Research
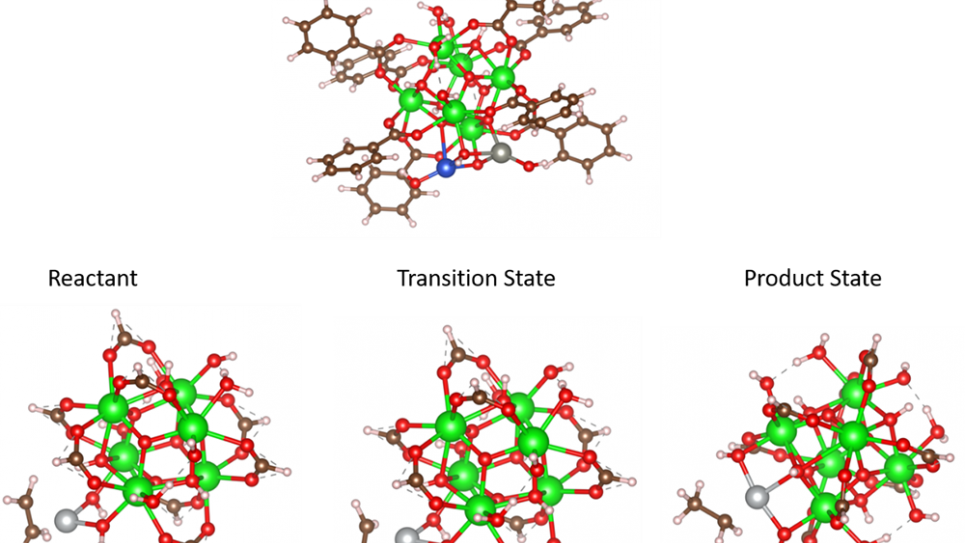
Quantum chemistry studies the ground state of individual atoms and molecules, the excited states, and the transition states that occur during chemical reactions. Quantum chemistry is required to measure the chemical reaction from high to low energy. Quantum simulations use the fundamental building blocks and compute the fundamental models that are important to real life and determine how electrons are reacting to the changes. Figure 1 shows the relative energies and chemical reactions during the Argonne research.
When doing molecular calculations, researchers typically use a basis set. Often a basis set size is chosen to reduce computer memory while reducing the accuracy of the calculation. Another way of doing calculations is to use plane waves which are very accurate because they use a full space basis set. However, calculating plane waves is extremely memory intensive.
For their metallorganic framework project, the researchers used plane waves to generate the molecules (up to 158 atoms) in huge boxes. For this system size, researchers typically would need approximately 950 Gigabytes (Gb) of memory. However, Dr. Ye Luo, assistant scientist at Argonne, implemented a new method to represent the data contained in 950 GB and reduce the size by a factor of ~7 without losing accuracy. This dropped the requirement to less than 160 GB of memory needed for the calculation. Benali states, “We used 1,000 Theta HPC system nodes for 12h per system (7 molecules) to reach the accuracies. It would not have been possible on other machines as we needed the large number of nodes and the large size of memory. The reason we used plane waves instead of other cheaper memory-wise methods, is because we were going after high accuracy that you cannot achieve with other methods.”
“Doing the plane wave calculation would normally require 1.2 terabytes of memory on a chip. The Argonne team did a hybrid representation that gave more accuracy while requiring only 146 gigabytes (GB) of memory. This was made possible by our quantum chemistry hybrid representation as well as the extra memory available on the Theta supercomputer. When Theta was constructed, a special feature added extra memory on the chip to aid in memory intensive calculations. Theta contains 192 gigabtyes of memory as compared to the average 64/128GB on a chip. Results of this new calculation will aid the broader scientific community that uses the metallorganic framework to determine the energy needed in catalysis used in applications such as semiconductors.”
Theta HPC System Advances Quantum Chemistry Research
Quantum chemistry research is currently performed on the ALCF Theta Cray X40 supercomputer. Theta, an 11.69-petaflops system, is a massively parallel, many-core system based on Intel Xeon Phi processors and interconnect technology, a new memory architecture, and a Lustre-based parallel file system, all integrated by Cray’s HPC software stack.
The Theta system is equipped with 4,392 nodes, each containing a 64-core processor with 16 gigabytes (GB) of high-bandwidth in-package memory (MCDRAM), 192 GB of DDR4 RAM, and a 128 GB solid state drive (SSD). Theta’s initial parallel file system is 10 petabytes. Theta has several features that allow scientific codes to achieve higher performance, including:
- High-bandwidth MCDRAM (300 - 450 GB/s depending on memory and cluster mode), with many applications running entirely in MCDRAM or using it effectively with DDR4 RAM
- Improved single thread performance
- Improved vectorization with Intel AVX-512
- Large total memory per node
Challenges for Future Quantum Chemistry Research
“Currently, the Argonne team is able to study systems containing up to 6,500 electrons with the Theta system. However, as the ALCF moves toward future Exascale computing with the upcoming Aurora HPC machine, they will be able to do 20,000 electrons. With the future Exascale Cray Aurora21 machine at ALCF, we anticipate being able to use quantum chemistry calculations for real, complex devices containing defects and interfaces. Exascale will make calculations at least 50 times faster,” indicates Benali.