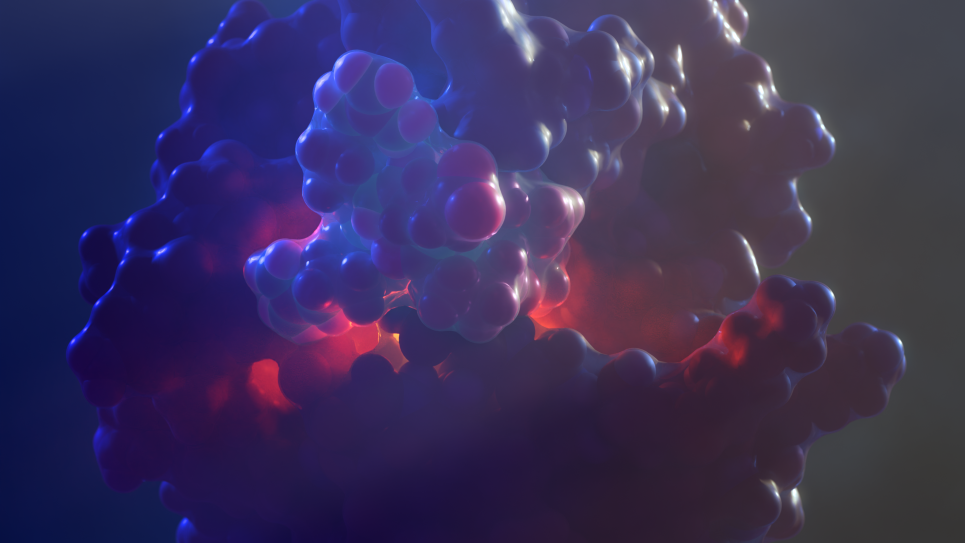
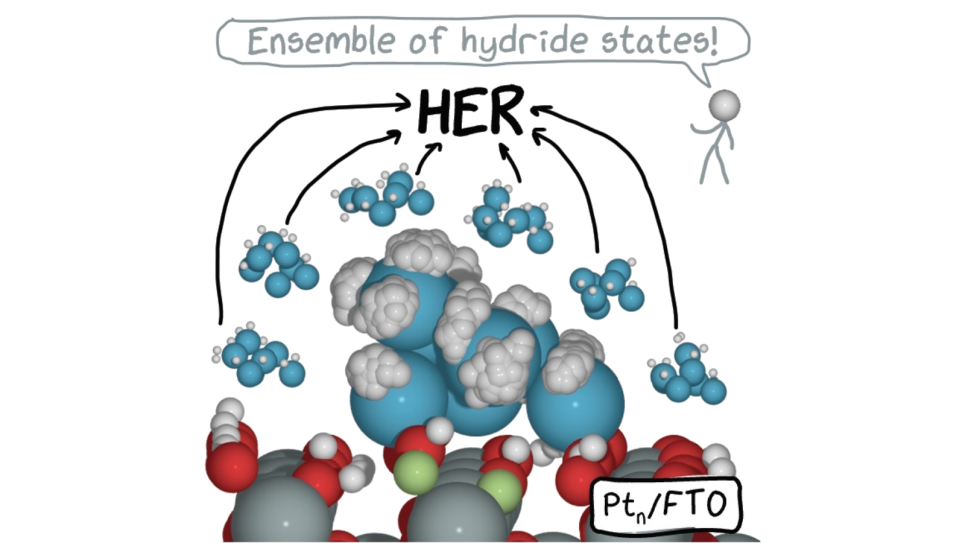
do not imply single rigid cluster isomer facilitating a single catalytic event in vacuum (B), but instead, realistic coverage, T, p, access to many cluster isomers (% in C indicating fractions of the population), and fluxionality, with activity being due to possibly not the most stable isomer.")
Dynamic Nanocluster Catalysis
Project Description
Today, heterogeneous catalysis is used in more than 90 percent of all chemical manufacturing processes. Catalytic materials enable selective formation of desired products at rates that are commercially viable. Catalysis is not only important for the production of chemicals, materials, and food, but also essential for pollution control, medical applications, and the development of sustainable energy solutions. Therefore, research on catalysis is a continuing developing area, which involves different sciences, such as chemistry, surface science, and materials science.
Small clusters of noble metals secured on supporting surfaces can be remarkable catalysts, with properties tunable through their size, nature of the support, and reaction conditions. The opportunities to improve the design of such catalysts are therefore vast. This INCITE project will use computation to understand and design the most powerful, selective, stable, and yet economical catalysts based on surface-mounted small clusters of transition metals. The exploration of such catalytic interfaces, which are incredibly complex and best represented as ensembles of many geometric states, requires the power and scale of DOE leadership-class supercomputers.
The research team will pursue the development and consolidation of protocols and efficient methods to enable the computational description of surface-mounted cluster catalysts in a statistical ensemble representation in realistic conditions. All the properties of cluster catalysts (e.g., reaction rates, stabilities, spectral signatures) will then be ensemble-averages, which can be used to explain existing experiments, or predict outcomes of new experiments.
Once the protocols and models are developed, the researchers will computationally manipulate the catalyst systems by varying temperature, partial pressures of gases, and cluster size to achieve the exposure of the most active binding sites and desired catalytic efficiency. The work will be done using ab initio methods and density functional theory (DFT) with and without periodic boundary conditions, as well as the team’s in-house methods for modeling materials in conditions relevant to their practical use, in conjunction with chemical bonding analysis tools. The team’s results will be used to inform experimental efforts to improve the performance of catalysts.