Towards Breakthroughs in Protein Structure Calculation and Design
Project Description
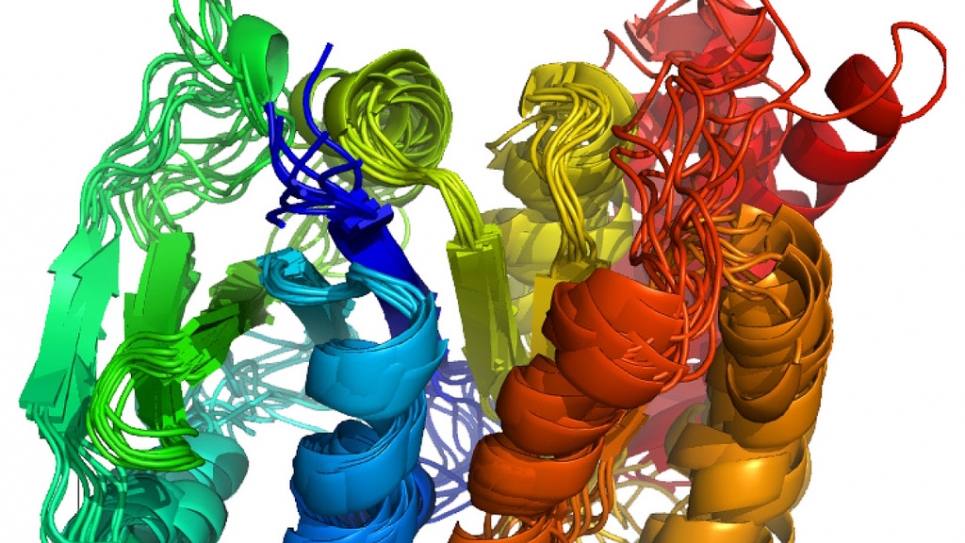
Proteins are the workhorse molecules of all biological systems. A deep and predictive understanding of life thus requires a detailed picture of their structure. Conventional protein structure determination using nuclear magnetic resonance (NMR) relies primarily on side-chain proton-proton distances. The necessary side-chain chemical shift assignment, however, is expensive and time-consuming, with possibilities for error. Moreover, approaches to NMR structure determination for larger proteins usually rely on extensive deuteration, which results in loss of key proton-proton distance information. Sparse structural data can be obtained from backbone-only experiments like orientational restraints from residual dipolar couplings and amid proton distances from NOESY spectra. These experiments are readily applicable even to fully deuterated and large proteins.
To determine NMR structures without side-chain chemical shift information, researchers incorporate backbone chemical shifts, residual dipolar couplings, and amide proton distances into the Rosetta high-resolution modeling methodology. To exploit the weak guidance signal provided by the sparse constraints, they developed an iterative scheme similar to a genetic optimization algorithm. A pool of the fittest individuals (e.g., lowest energy conformations) is maintained, and its worst part is replaced with offspring. The breeding or crossover of highly fit species (e.g., low energy conformations) is implemented as a Monte Carlo optimization that recombines features of previously found low-energy conformations. The type of features selected for recombination is adapted to the resolution of the pooled low-energy structures.
The iterative protocol increased the size range of accessible protein structures compared to the conventional Rosetta protocol. Researchers consistently solve protein structures up to 200 residues. Currently, they are determining the size range of this method and are testing further improvements. The INCITE program has been and will continue to be invaluable in its development.
Our INCITE work is focused on three areas currently. The first area is computing protein structures from very limited experimental data. With the INCITE computing resources, we are optimistic about developing methods which allow determination of the structures of proteins over 200 amino acids by NMR, which would be a big breakthrough in this area. The second area is designing proteins to bind very tightly to specific regions on a specified target. The third area is design of new enzyme catalysts. We are exploring catalysts for hydrogen production, solar capture, and other energy-related applications.
Domains
Allocations