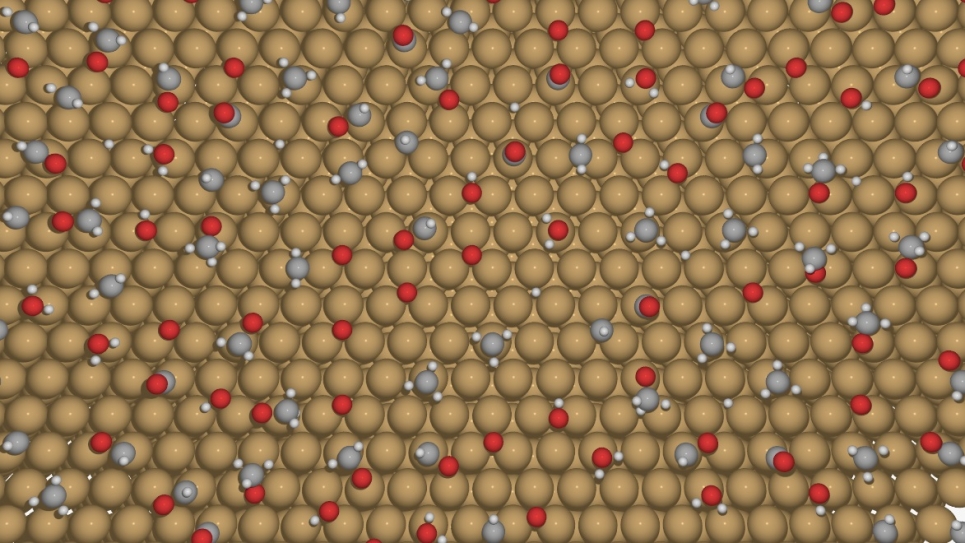
The SIPS approach to solving the eigenproblem

Advances in computing power are revolutionizing the field of chemical synthesis, which seeks to create new materials and chemicals. Synthesis is achieved by breaking and forming chemical bonds in a specific sequence, and, depending upon available starting materials and the desired end product, may involve a considerable number of individual reactions.
An Argonne National Laboratory team of scientists is developing methods and software to predict the reaction thermodynamics and kinetics in synthesis processes. The team’s work is expected to enable the discovery and optimization of synthetic approaches to valuable products, such as novel photovoltaic materials, and the inhibition of undesirable products, such as soot arising from combustion.
“Our work is really about synthesis and could apply to solution chemistry and materials chemistry,” said Albert F. Wagner, the Argonne Distinguished Fellow who leads the team. Other team members include Argonne postdoctoral researcher Murat Keçeli and physicist Peter Zapol, University of Alabama Robert Ramsay Professor of Chemistry David A. Dixon, and Argonne computational scientists Hong Zhang and Alvaro Vazquez-Mayagoitia.
Synthesis is all about breaking and forming chemical bonds, which result when two different atoms share their electrons. Electrons can only be reliably described by quantum mechanics, which can determine their energies (characterized by eigenvalues) and their spatial distribution (characterized by eigenvectors).
The numerical solution of the eigenproblem for the eigenvalues and eigenvectors is generally the most time-consuming part of computing synthesis mechanisms. Finding the solution to an eigenproblem for tens to tens of thousands of atoms rapidly escalates to millions of variables and equations that can only be handled by the power of a supercomputer.
The team successfully tested an approach to harnessing the massively parallel architecture of the Argonne Leadership Computing Facility’s (ALCF’s) supercomputer, Mira, to solve this part of the computation, opening the door for significant future research campaigns. The ALCF is a U.S. Department of Energy Office of Science User Facility.
Materials science investigations typically favor one of two modeling approaches: atomistic (microscale) or continuum (macroscale). The choice of which method to use is essentially a trade-off between precision and feasibility. Yet this gap between scales could, in principle, be narrowed or eliminated with reduced scale approaches that make use of the locality of chemical interactions and advanced computational techniques.
Density-functional based tight-binding (DFTB) is one such approach and can produce useful molecular structures and energetics at a significantly reduced computational cost. Researchers can simulate thousands of atoms undergoing bond breaking and forming synthesis processes with acceptable accuracy.
Unfortunately, these methods (and others) have a major bottleneck in the solution of the eigenproblem via the diagonalization of a key matrix. Many problems can be represented with matrices, and while supercomputers excel at manipulating matrices, this diagonalization step is not easily scalable to parallel computing. Various characteristics of a matrix, such as whether it is sparse or dense, must be used by a solver to efficiently divide the problem into independent tasks that can be done in parallel by a supercomputer’s nodes.
The Argonne team needed a way to test a novel approach for solving the eigenproblem for reduced-scale electronic structure methods applied to large systems that could enable future research at a larger scale. This approach is called SIPs, which stands for shift-and-invert parallel spectral transformations. SIPs was initially developed at Argonne in 2007 by Zhang, Zapol, Barry Smith, and Michael Sternberg to efficiently solve the eigenproblem and thereby obtain the eigenvalues and eigenvectors. The current team optimized the method for massively parallel architectures and specifically enabled DFTB calculations for systems with more than 100,000 atoms utilizing more than 200,000 CPU cores.
In early 2015, the Argonne team was given a Director’s Discretionary allocation on Vesta, ALCF’s test and development platform, and on Mira, its 10-petaflops Blue Gene/Q, to test the robustness and scaling capability of their approach. They demonstrated that SIPs could scale beyond 200,000 Blue Gene/Q cores and solve more than 300,000 eigenpairs of a 500,000 by 500,000 matrix in approximately three minutes.
The team developed a Python code, PSCF, for faster prototyping the solver for different quantum chemistry methods with very good scaling performance. The team is also working on the integration of SIPs with widely used electronic structure packages MOPAC and SIESTA, and testing the integrated software on benchmark systems, such as water clusters, graphene sheet, and large biomolecules. The team ran these jobs on 4,096, 8,192, 16,384, 32,768 and 49,152 nodes to obtain scaling data for a future INCITE proposal for more computing time.
SIPs has been implemented through PETSc, a well-known Argonne-developed suite of data structures and routines for the scalable solution of scientific applications modeled by partial differential equations. SIPs also depends on the non-Argonne SLEPc library, which is designed for solving large-scale sparse eigenvalue problems. (SLEPc now also hosts a recent implementation of SIPs.) The Argonne team is working with SLEPc developers to further tune this solver for quantum chemistry calculations.
ALCF’s Vazquez-Mayagoitia, a computational and theoretical chemist himself, helped the team to port and interface the code with the quantum chemistry packages and provided advice on eigensolver algorithms. He also assisted with the performance tools used to scale the codes on Mira and held technical discussions regarding similar techniques on petascale architectures.
Now with the availability of a scalable implementation to diagonalize matrices using a shift-and-invert eigenvalue algorithm, it is possible to simulate hundreds of thousands of atoms using hundreds of thousands of processors.
A paper on SIPs was published in the Journal of Computational Chemistry.
M. Keçeli, H. Zhang, P. Zapol, D. A. Dixon, A. F. Wagner. J. Comput. Chem. 2016, 37,448–459. DOI: 10.1002/jcc.24254.
This research is supported by DOE’s Office of Science. Computing time at the ALCF was allocated through ALCF’s Director’s Discretionary program.
Argonne National Laboratory seeks solutions to pressing national problems in science and technology. The nation's first national laboratory, Argonne conducts leading-edge basic and applied scientific research in virtually every scientific discipline. Argonne researchers work closely with researchers from hundreds of companies, universities, and federal, state and municipal agencies to help them solve their specific problems, advance America's scientific leadership and prepare the nation for a better future. With employees from more than 60 nations, Argonne is managed by UChicago Argonne, LLC for the U.S. Department of Energy's Office of Science.
The U.S. Department of Energy's Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, visit the Office of Science website.
Domains