Software optimized on Mira advances design of mini-proteins for medicines, materials
Scientists at the University of Washington are using Mira to virtually design unique, artificial peptides, or short proteins. Peptides have the best properties of two different classes of medical drugs today and could enable future, peptide-based medicines with few side effects. As researchers begin to develop new peptides, they are optimizing their in-house software to test thousands of potential peptide structure designs in tandem, requiring a state-of-the-art supercomputer.
When diseases like cancer and AIDS affect millions of people each year and rare but deadly infections like Ebola move faster than treatment, the race to make effective drugs becomes a sprint.
That's why University of Washington (UW) researchers are using one of the nation's most powerful supercomputers, the 10-petaflop/s Mira at the Argonne Leadership Computing Facility (ALCF), to improve their software for designing protein structures to likewise virtually design and test mini-proteins called peptides. The ALCF is a U.S. Department of Energy (DOE) Office of Science User Facility.
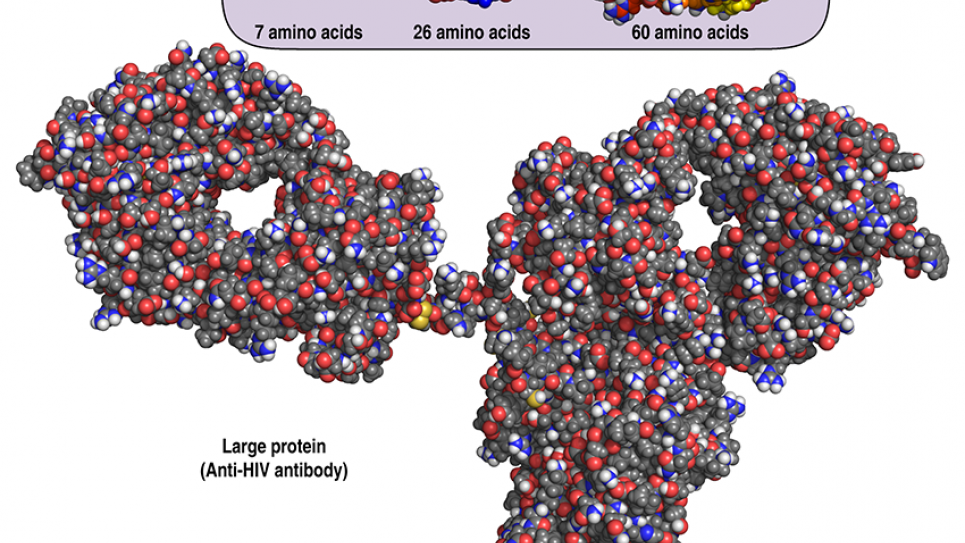
Peptides are made up of chains of amino acids — the 20 basic organic compounds found in proteins — but they are about one-tenth the size of proteins. Peptides can bind to targets on a cell (usually receptors or enzymes that are themselves proteins) and so regulate cellular functions such as nutrient uptake or communication between cells. A peptide's structure, including how the branches of its amino acids fold into three-dimensional conformations, determines which targets it can "fit" or bind to.
By computationally simulating and testing peptides in large numbers, the UW Baker Laboratory, led by principal investigator David Baker, envisions designing peptides that have unique structures not found in nature. These "designer" peptides could one day make up a new class of therapeutic drugs that is more effective and results in fewer side effects. They could also have widespread applications in technologies and materials that rely on organic compounds, such as self-assembling or super-strong materials.
A good fit
Many drugs today are "small-molecule" drugs in which molecules of active ingredients easily dissolve and diffuse because they are small enough to cross key membranes in the body.
"Most drugs are administered systemically (through the circulatory system), even if they need to go to only one place in the body, which means they have to be administered in higher doses," said Vikram Mulligan, UW senior fellow and co-principal investigator. "When small molecules are administered, they can stick to many different proteins in the body because their size enables them to interact with many targets, not just the intended target, causing more side effects."
Mulligan cited chemotherapy as a good example of this effect: "When you're on chemo, you're quite sick because the drug is affecting your whole body, not just the cancer."
For more than 20 years, the Baker Laboratory has developed computational modeling tools for protein design, which is needed for making targeted protein therapies that reduce side effects. Using the Baker Laboratory's flagship protein structure and sequencing software, Rosetta, researchers can predict protein structure from amino acid sequences and design new amino acid sequences to yield a desired function.
However, while small-molecule drugs can cause unwanted side effects because they are easily absorbed in the body, protein drugs can have the opposite problem: they can be too big.
"The advantage of the protein is that it has great efficacy and is likely to produce the desired result," Mulligan said, "The disadvantage is that proteins are large. They're hard to get into the body, and they have difficulty crossing membranes in the body. On top of that, we have an immune system that is good at recognizing foreign proteins and eliminating them."
Peptides strike a promising compromise, but in order to make these short "floppy" chains of amino acids into distinct rigid shapes that can be customized for specific targets, researchers have to reinvent their amino acid building blocks. Alongside the Rosetta "computational lab," Baker Laboratory scientists can synthesize peptides from new artificial amino acids in their physical wet lab as well as test their final protein and peptide designs.
By introducing artificial amino acids into peptide simulations, the number of potential uniquely shaped peptides skyrockets. Using artificial amino acids also offers some protection against the immune system, which might otherwise recognize natural amino acid sequences as foreign proteins and attack them.
A new chapter for Rosetta
Over the last half-century, protein structure data from imaging techniques such as X-ray diffraction and electron microscopy has mounted, and protein structure databases store at-the-ready information on sequencing and structure.
However, researchers are still trying to thoroughly understand how sequence influences structure, particularly folding. In order to find the best protein or peptide sequence for a specific function, they must simulate thousands of designs and search for the most effective option. This optimization search requires introducing a range of geometric constraints at the atomic level that could influence folding and binding. The entire space for a protein or peptide structure's dynamics is called its conformational space.
"This is an exhaustive approach," said Yuri Alexeev, ALCF assistant computational scientist. "To model the conformational space for peptides requires hundreds of millions of simulations."
To simulate a new peptide, Rosetta begins with a single-state design. Working backward from the target on a virus, malignant cell or other disease-causing agent, researchers determine what the basic conformation of the peptide should be. They use this peptide "skeleton" to generate thousands (or even hundreds of thousands) of possible amino acid sequences that could support stable interactions for that conformation. These first steps can be carried out on smaller systems because the potential sequences can be generated in isolation.
However, evaluating and optimizing these thousands of designs requires rapid communication between processing cores, which is why Mira — and its more than 780,000 cores — is needed.
To eliminate side effects, researchers not only want a peptide that favors, or is stable in, the conformation that will effectively bind to a desired target, they also want a peptide that disfavors any other conformations so there is little chance it will change conformation and interact with other proteins in healthy cells, causing side effects.
Through a multistate design algorithm, Rosetta simulates and optimizes each conformational state on a separate core and searches for the state that favors only the desired conformation.
The Baker Lab originally developed the multistate design algorithm for protein design, but it needs fine-tuning to tackle smaller peptides. Because Rosetta uses potential energy functions to calculate the interactions between atoms and molecules in a protein, the team is improving the energy function to allow it to be better applied to systems that include new amino acids for which experimental data might not be available. This step, unique to peptides, requires developing energy functions that not only use existing data from natural proteins to approximate interactions but also use the laws of physics, adding to the computational challenge.
"If we're designing a large protein and we have some mistake or inaccuracy in our energy function, there's a good chance it will average itself out over the large structure," Mulligan said. "However, with a peptide, we're considering a smaller number of interactions and an error might throw our design off quite a bit, so our energy function has to be more precise."
In 2012, the Baker Laboratory wanted to transition Rosetta, focused on protein design at the time, from the facility's previous Blue Gene/P system to Mira, a Blue Gene/Q system. Alexeev helped port and benchmark Rosetta for the Blue Gene/Q and advised on how to increase parallelization. Computing time for the peptide work was provided through a 2014–2015 Advanced Scientific Computing Research Leadership Computing Challenge award and a 2015 Innovative and Novel Computational Impact on Theory and Experiment (INCITE) award. Funding for research was provided by the National Institutes of Health National Institute of Aging and the National Science Foundation.
Since the start of the team's most recent INCITE project, Alexeev has also been helping validate a force field (the sets of parameters needed to accurately calculate the energy functions) for peptide design against quantum-mechanical simulations. This force field will greatly improve predictions for designs using artificial amino acids.
Seeking targets for peptide design, the team has begun preliminary investigations into proteins on the surface of the HIV capsid and Ebola and Marburg viruses, as well as histones (also proteins) involved in some types of cancer.
In the long run, they hope to assemble a database of artificial peptide designs that will provide Rosetta users with a scaffold, or template, to cut down on the computational effort needed to optimize a peptide for a medicine or material application.
"Such a database would help future design efforts because, for example, once we have that, we can pick out a peptide design that serves as a scaffold to be redesigned and optimized for a new disease, rather than designed from scratch," Mulligan said.
Argonne National Laboratory seeks solutions to pressing national problems in science and technology. The nation's first national laboratory, Argonne conducts leading-edge basic and applied scientific research in virtually every scientific discipline. Argonne researchers work closely with researchers from hundreds of companies, universities, and federal, state and municipal agencies to help them solve their specific problems, advance America's scientific leadership and prepare the nation for a better future. With employees from more than 60 nations, Argonne is managed by UChicago Argonne, LLC for the U.S. Department of Energy's Office of Science.
The U.S. Department of Energy's Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, visit the Office of Science website.