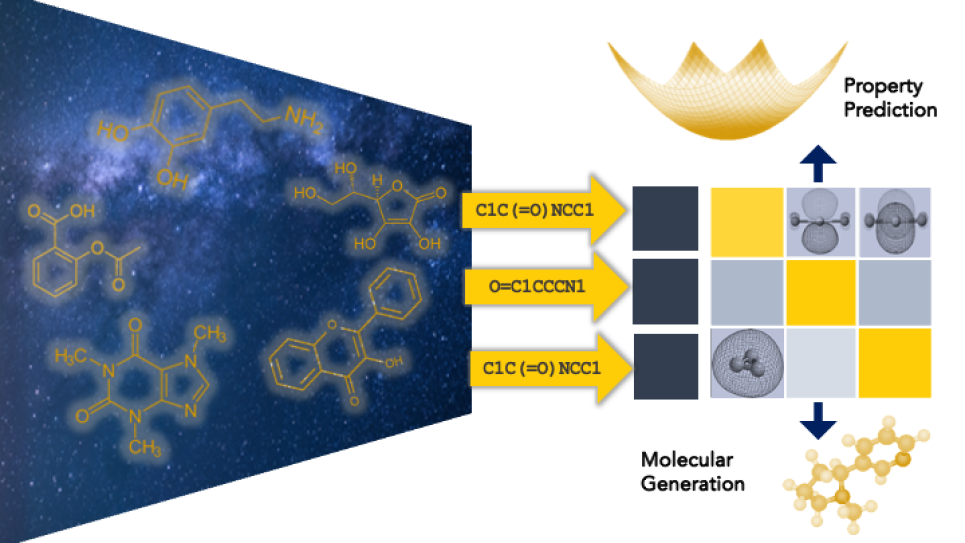
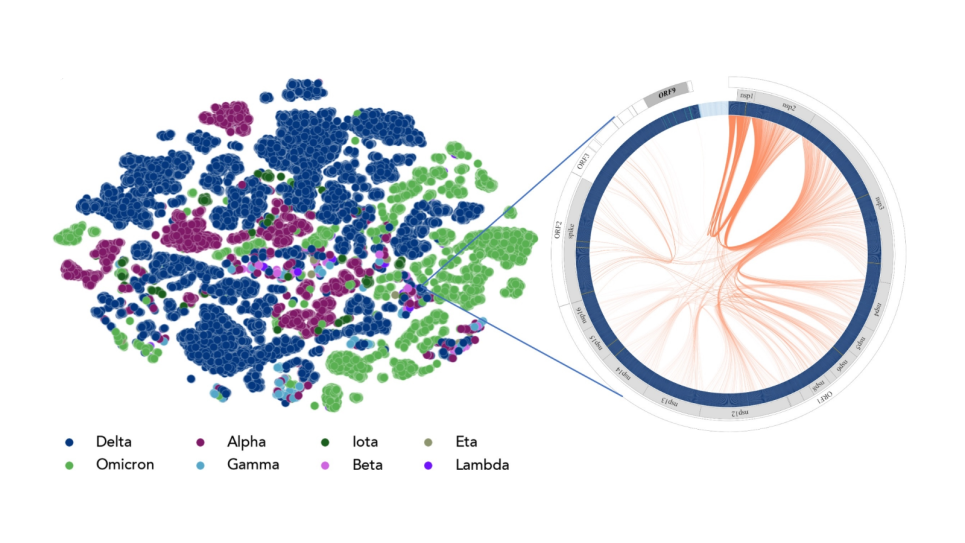
Protein-Protein Binding Specificity
Project Description
Protein-protein interactions are ubiquitous in many biological pathways and play an essential role in modulating cellular functions. However, computing the binding affinity of even two proteins to determine whether they will associate and form a stable complex is a technical challenge. This project aims to understand and predict protein-protein associations that govern biological processes at the atomic level using a molecular dynamics (MD) computational strategy, based on computationally intensive, all-atom models. Researchers are validating this computational method against a broad range of protein complexes to see if binding affinity can be predicted with quantitative accuracy.
Using NAMD scalable molecular dynamics software, the research team has developed a powerful and scalable computational method that breaks down binding calculations into several steps. It then expresses the binding free energy as a sum of free energies associated with each step that can be calculated from the replica-exchange MD (REMD) potential of mean force calculations, a type of energy change calculation. In REMD simulations conducted on Mira, an ensemble of replicas of the protein-protein interaction that cover a range of conditions is considered. This method increases the sampling efficacy of MD simulations.
The team is quantifying the binding affinity and binding entropy for several characteristic protein complexes, including cohesin-dockerin proteins that control the assembly of the cellulosome (enzyme complexes that facilitate the decomposition of plant cell walls) and are, therefore, important for energy production. By studying complexes of various size and flexibility, the team is advancing theory-modeling-simulation (TMS) technology. With an established TMS route to predicting protein-protein interactions, years of high-resolution structural data collected in large protein interactome databases could be leveraged to address fundamental biological questions and help solve the problem of rational protein design.
Domains
Allocations