
Accelerated Catalyst Discovery. Image: Rajeev Surendran Assary, Argonne National Laboratory
Accelerated Catalyst Discovery. Image: Rajeev Surendran Assary, Argonne National Laboratory
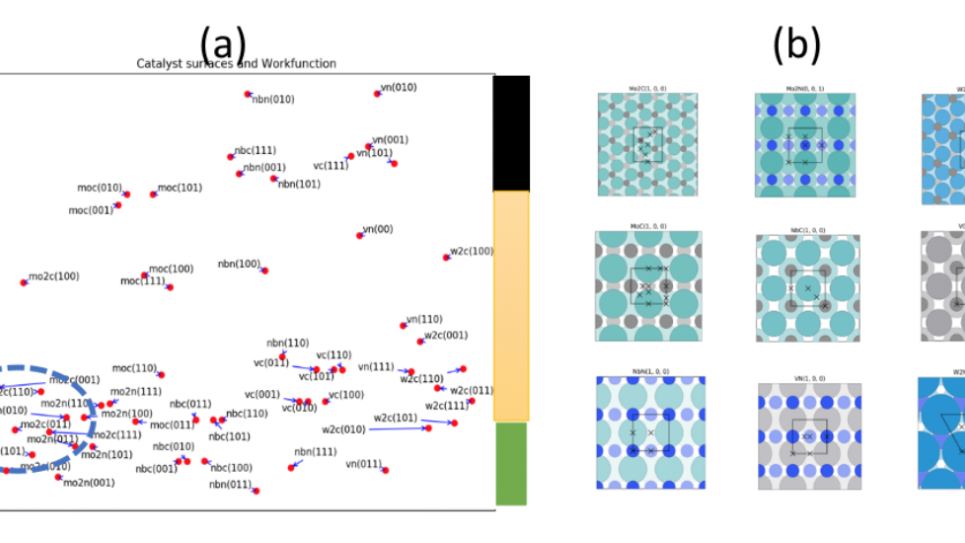
This project will utilize density functional theory and machine learning methods that enable exhaustive searches for active catalyst facets (monometallic and bimetallic) and reveal active site motifs for deoxygenation and C-C bond formation reactions.
Catalysts enable faster chemical transformation of all industrial processes essential for humankind. New and improved catalysts are desired for converting naturally abundant non-edible biomass and other waste streams to value added chemicals and transportation fuels. The crucial bottleneck associated with the catalyst discovery process is the cost and lack of a priori understanding of the properties of the catalyst when interacting with chemicals or reaction intermediates. In particular, chemical transformation such as deoxygenation (removal of oxygen atoms) and the carbon-carbon (C-C) bond formation reaction of bio oil components are essential to produce desired candidates for transportation fuels, are critical reactions that require efficient and durable catalysts. Here we propose to develop and utilize an automated computational approach that utilizing high fidelity first principles simulations coupled with machine learning (ML) to provide guidelines to the catalyst discovery challenges.
This project will utilize density functional theory (DFT) and ML methods that enable exhaustive searches for active catalyst facets (monometallic and bimetallic) and reveal active site motifs for deoxygenation and C-C bond formation reactions. Utilization of massively parallel computational architectures to generate first principle catalyst data along with an active learning approach would provide a unique toolset to search economically and synthetically accessible ‘catalyst subspace’ (Metal carbides, nitrides, and phosphides) that enable the discovery of most promising catalysts. This catalyst space and their selected alloys will be investigated and the guidelines and the design of catalyst candidates obtained from the investigation will be feed into the discovery/verification efforts of ChemCatBio consortium (https://www.chemcatbio.org/), which is part of the Energy Materials Network funded by the Energy Efficiency and Renewable Energy (EERE), Bio Energy Technology Office (BETO).


