Free Energy Landscapes of Membrane Transport Proteins
Project Description
Tier 1 Science Project
Science
Membrane transport proteins are a unique class of macromolecular biological systems that play a critical role in numerous aspects of cell function. A distinct feature of those molecular processes is the existence of large conformational changes, often in response to, or in concert with, the surrounding chemical environment. A complete understanding of how these proteins carry out their function will thus rely heavily on characterizing these transitions. However, such conformational pathways are inherently difficult to determine directly from experiment due to the involvement of transient, short-lived states. Progress on this front will ultimately be possible only by maintaining a strong link between computational and experimental efforts.
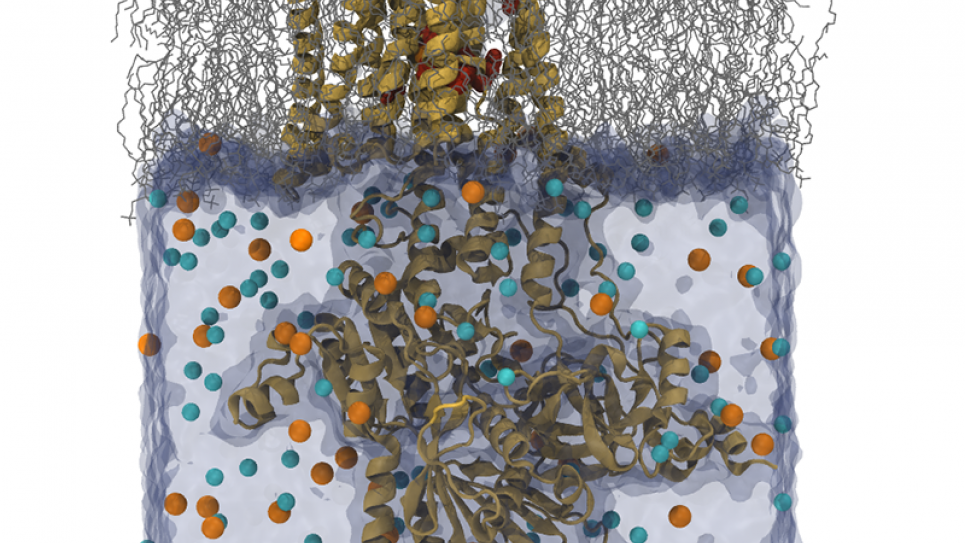
The main interest here will be to determine the free energy landscapes of large membrane proteins, especially P-type (e.g., the Na+/K+ pump) and F/V-type ATPases (e.g., the Fo/Vo unit of ATP synthase). In this project, researchers will use atomistic computational models to determine the free energy landscape of large-scale conformational transitions for both the NaK pump and Fo-ATPase. The necessary simulations will build upon existing structural data from crystallography and provide meaningful connections to measurements on real, dynamical systems. Missing structural data will naturally be predicted by string method simulations and subsequently characterized to assess validity and potential importance for experimental observations.
A large array of simulation techniques will be used on this ambitious project. While long-standing methods, such as umbrella sampling and replica exchange, are now staples of the biomolecular simulation community, newer approaches, such as string simulations with swarms of trajectories and hybrid MD/Monte Carlo constant pH protocols, will also be employed.
Impact: An atomistic picture of membrane transport proteins is a critical component for understanding a broad range of biological functions. This work will utilize computational models to provide both detailed visualizations of large protein motions as well as quantitative predictions into the energetics of these processes.
Problem Scale
The NaK pump and Fo-ATPase proteins are transmembrane proteins with large cytoplasmic domains. A typical simulation size for these proteins is more than 150,000 atoms. Researchers expect to perform string method calculation for initial pathway identification and equilibration with all-atom detail for at least 50 ns of simulation; resource estimates based on using 64 images and 16 replica per images for string method calculation. For preparation of the free energy simulation, the team needs to perform equilibration of 50 ns for each replica; resource estimates based on 256 replicas being used in the free energy simulation.
Numerical Methods/Algorithms
NAMD supports classical molecular dynamics simulations, most commonly of all-atom models with explicit solvent and periodic boundary conditions with PME full electrostatics in an NPT ensemble, although coarse-grained models, implicit solvent, and non-periodic or semi-periodic boundary conditions are also supported. CHARMM and similar force fields are supported, including the Drude polarizability model. Also supported are alchemical free energy calculations and methods for accelerating sampling, including user-customizable multiple-copy algorithms for parallel tempering and conformational or alchemical free energy calculations
Parallelization
NAMD is parallelized using Charm++, an overdecomposition-based message-driven parallel programming model. Charm++ encourages decomposition of the application domain into logical entities that resemble the data and work units of the application. The program is defined in terms of these entities, encapsulated as C++ objects, called chares. Under the hood, a powerful runtime system (RTS) maps and schedules execution of the chares on processors. On most machines, Charm++ is implemented on top of the low-level communication layer provided by the system, for best performance (called native builds); on Blue Gene/Q, it is implemented on PAMI.
Application Development
- The team’s overarching optimization strategy will be a transformation of the way NAMD is mapped onto the increasingly many Charm++ worker threads within a process from thread-centric, representing an aggregation of otherwise distinct streams of execution, to process-centric, dynamically scheduling available work to minimize latency or maximize throughput as needed. Process-centric programming is being prototyped and adopted incrementally to address performance bottlenecks within the current NAMD codebase.
- NAMD will use at most 4 Charm++ processes (equivalent to MPI ranks) per node, with whatever number of threads per core gives the best-measured performance. The communication threads would each have a dedicated core while the worker threads would be 2-4 per core.
- The proposed simulations should fit easily in 16GB of MCDRAM per node, so the appropriate MCDRAM usage model will be used.
- NAMD’s collective variable module is an essential toolkit for modern simulation techniques, but complex collective variables involving hundreds of atoms can present a serial bottleneck. The team will parallelize (or partly parallelize) the collective variable module in NAMD.
- Researchers will implement a new hybrid algorithm that rigorously combines Monte Carlo and MD simulations to significantly increase conformational sampling as well as to allow simulation at more complicated conditions such as constant pH. They will incorporate this into NAMD to compute potentials of mean force of transport molecules across membranes
Portability
NAMD development has been specifically geared towards addressing both high-end and low-end platforms, with and without NVIDIA GPUs or Intel Xeon Phi co-processors, and with x86 64, POWER, ARM, or self-hosted Xeon Phi CPUs. The researchers do not see any future alternative for programming NVIDIA GPUs that will be as well-supported as CUDA, but OpenMP 4.0 SIMD directives provide at last a standard cross-platform means of reliably accessing vector instructions. These SIMD directives will be most critical on non-GPU- accelerated machines, which will predominantly have x86 64 and/or Xeon Phi processors, both targets of the Intel compiler. Hence, the team will first write OpenMP 4.0 SIMD kernels vectorizable by the Intel compiler, and only later address ARM and POWER performance for those kernels that are not offloaded to the GPU via CUDA. They will use identical data structures between Xeon Phi CPUs and offload coprocessors, and also other CPUs and GPUs whenever reasonable, using modern C++ features to keep as much code in common as possible.
The portability of NAMD between different networks is already handled by Charm++, which has optimized network layers for every major modern network type. However, should a new network API arise, the researchers would, rather than targeting it directly, instead create a Charm++ GASnet network layer in the expectation that GASnet itself would then be ported to any other future network APIs. They will also explore dropping Intel offload directives and instead using Charm++ to schedule directly on the Xeon Phi coprocessor.
Domains
Allocations