
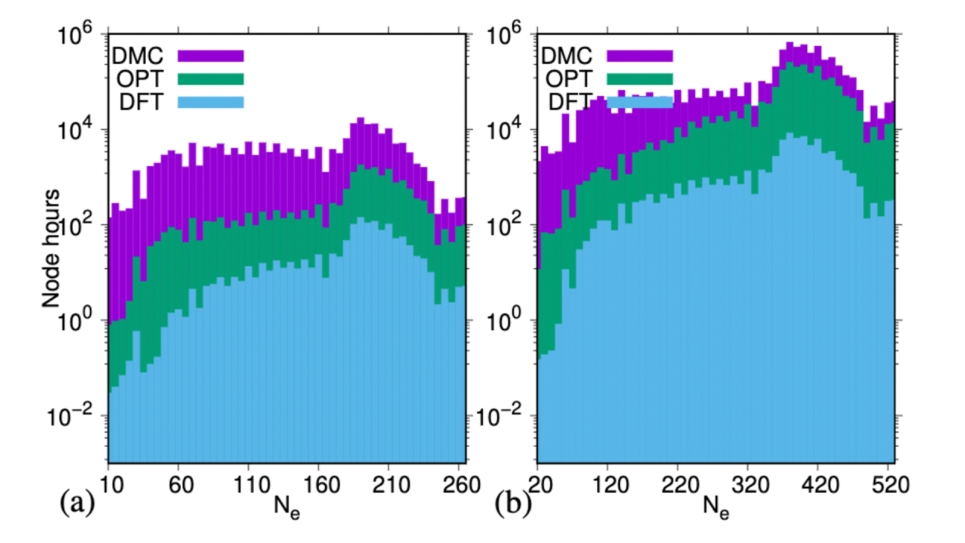
Computational cost of DFT (Hybrid functional), Jastrow opti-mization, and DMC calculations for each group of conformers (a) with afixed number of electrons, and for the corresponding group of dimers (b).For each dimer we consider 11 structures.
Computational cost of DFT (Hybrid functional), Jastrow opti-mization, and DMC calculations for each group of conformers (a) with afixed number of electrons, and for the corresponding group of dimers (b).For each dimer we consider 11 structures.
This project aims to create a new database of non-covalent interactions, in and out of equilibrium with a combination of density functional theory (DFT) with many-body-dispersion method (DFT+MBD) and the diffusion Monte Carlo (DMC) method.
In the design of advanced molecular materials and in the discovery of new drugs, it is crucial to explore in an accurate and extensive way the chemical compound space—the phase space containing all feasible molecular compositions and conformations. Moreover, the understanding of how the various chemical and physical properties behave in and out-of-equilibrium is fundamental to reconstruct possible interaction patterns and the formation paths of stable conformers.
In this regard, non-covalent van der Waals (vdW) interactions play a crucial role for the qualitatively correct and quantitatively accurate description of not only the binding processes, but also of the structural, kinetic and spectroscopic, properties of an extensive set of molecules and materials. While there have been many attempts to model these interactions and overcome prohibitively high computational costs, the vdW models rely on accurate benchmark calculations. Moreover, in order to improve vdW models, reference calculations must be extended to large supramolecular complexes with an increase of the computational cost.
This project aims to create a new database of non-covalent interactions, in and out of equilibrium with a combination of density functional theory (DFT) with many-body-dispersion method (DFT+MBD) and the diffusion Monte Carlo (DMC) method. The database will include an accurate evaluation of the interaction energies via diffusion Monte Carlo (DMC). Through DMC calculations the researchers will be able to obtain extremely accurate interaction energies with a relative error of approximately 5%, overcoming the computational limitations that arise in traditional high-level quantum chemistry methods. This work will provide reference data for more accurate vdW models, and will be able to establish for the first time an extendable procedure to generate highly accurate databases via a combined DFT+MBD and DMC workflow.


