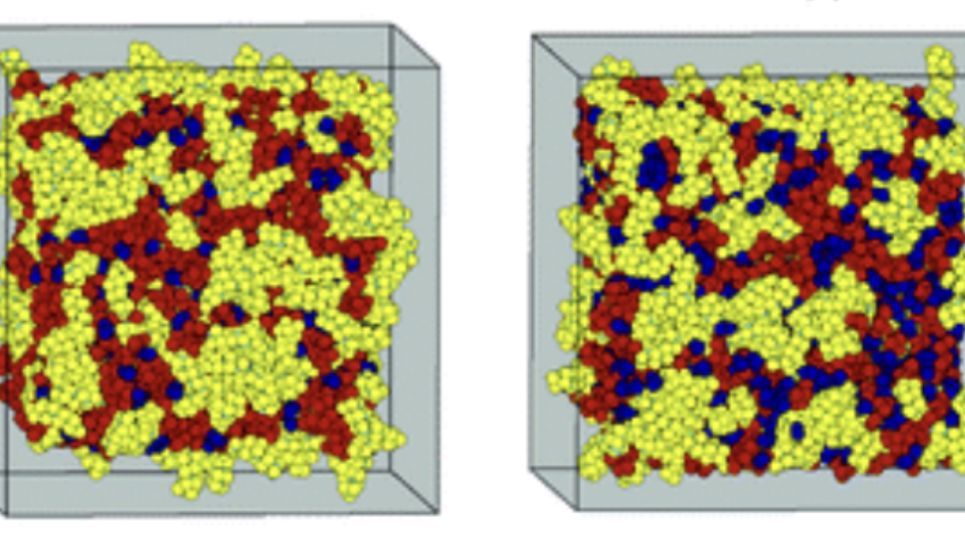
Nanostructural organization of ionic liquid (yellow & red) under varying additives (blue).
Nanostructural organization of ionic liquid (yellow & red) under varying additives (blue).
There is an increasing worldwide demand for high energy density batteries. This research is aimed at using large-scale, high performance computing to assist the discovery of novel battery electrolytes.
The exploration of new Li-ion battery materials is an important focus of materials scientists and computational physicists and chemists throughout the world. The practical applications of Li-ion batteries and emerging alternatives may not be limited to portable electronic devices, and circumventing hurdles to their widespread adoption in electrical vehicle applications, requires new electrode materials and a fuller understanding of how the materials and the electrolyte chemistries behave. Computational prediction of ideal is a leading methodology in designing materials and electrolytes optimized for function, including those for Li-ion batteries.
The planned computation constitutes the simulation part of the current EERE VTO project, nontraditional electrolyte design from ionic liquid. The overall goal is to enable rational design of superior electrolytes for high voltage batteries. This study will focus on nontraditional electrolyte discovery from ionic liquids: a new entry to battery electrolytes. The influence of structural perturbation at electrolyte molecules, such as fluoridation of cation ring, on nanostructural organization at electrolyte/electrode interface as well as the transport properties and desolvation/solvation kinetics of charge carriers will be examined with advanced computational methodologies, focused on exploring an optimal structure perturbation (synthesis) path to improve electrolyte performance in lithium ion transport. The high-throughput capability will allow use of characterization approaches from simulation studies to link solution correlations with influences on lithium ion-transport behavior in electrolytes and enable the ability to seek multiscale structural attributes that allow facile and selective incorporation of the charge carrier while prohibiting the dissolution of cathodic transition-metal components.
This project is focused solely on computational methodologies that benefit from using preexascale supercomputers, decreasing time to solution from months to days. Molecular dynamics methodologies such as Hamiltonian Annealing and sampling enhanced free energy calculations are ideal both for the research problems described here and the computer resources available for the allocation. Overall, this research will enable enhanced, fundamental understanding of how the charge carriers transport in hierarchical structuring of electrolytes and how simulation knowledge can be transferred to chemical synthesis and industrial environments. Advances enabled by this work will aid in the development of the US battery industry.