The search for new drugs to treat diseases is usually a long and arduous process. From initial investigation to drugs hitting the market, the process for anti-viral drug discovery can take 10 to 15 years and billions of dollars. From the beginning of the COVID-19 pandemic the question loomed: how long would it be before there were drugs that could treat the disease?
The pandemic has applied urgent pressure to make this process faster and more efficient. In the past few years, scientists have been using artificial intelligence (AI) and supercomputing resources, including those at the Argonne Leadership Computing Facility (ALCF), to streamline the process, combining different methods of drug candidate analysis to drastically reduce the time to drug discovery. The ALCF is a U.S. Department of Energy (DOE) user facility at DOE’s Argonne National Laboratory.
Identifying molecules that can bind to target proteins on viruses such as SARS-CoV-2 is laborious and time-consuming because the number of possible molecules is astronomical. There are billions of compounds to be sorted through to see if they can bind to the target proteins.
“When you have billions of possibilities, you have to narrow them down to a few thousand before you can proceed with these procedures, it is a huge task, and it is tedious,” says Agastya Bhati, a researcher from University College London who is using the ALCF’s Theta supercomputer for a project to combine protein binding analysis methods.
Faced with this huge and tedious task, the team sought a way to speed it up by using different methods to overcome the shortcomings they would run into if they used each method individually. “The biggest ambition,” Peter Coveney, a professor at University College London leading the project, says, “relates to the conflation or joining of machine learning methods to accelerate discovery through first looking at huge numbers of potential compounds and then ranking them in a way that’s prioritized based on increasingly accurate calculations.”
The traditional process for determining which compounds can bind to a target protein is to use physics-based simulations to calculate and rank how well each compound can bind. There are many physics-based methods with different degrees of computational intensity. At the beginning of the process, researchers start with less expensive, or less computationally intensive, methods to narrow the field of molecules to sift through. “You use very cheap methods, which are also not that accurate, but then you just discard a lot more molecules, so you have a smaller percentage of molecules and then you use progressively more accurate and more computationally intensive models, and it goes on like that,” Bhati explains. The drawback of this process is that as it becomes more accurate, it becomes more computationally intensive and takes a long time.
In a paper published in the Journal of Chemical Theory and Computation, the team detailed the use of one of the physics-based methods in this process, the Thermodynamic Integration (TI) method. TI is one of the most accurate and computationally expensive methods, so it is not used until the very last stages of the process, when the list of compounds is very short. A derivative of the TI method, developed by Bhati to make TI more statistically accurate and precise, is the Thermodynamic Integration with Enhanced Sampling (TIES) method. TIES runs ensemble simulations, or multiple replicas of simulations. The team has demonstrated its accuracy and reliability, but it can only rank compounds that are chemically similar to each other, so it is used toward the very end of the process. Once structures with potential have been identified, they are varied slightly to try to improve binding with the protein. This is known as the lead optimization stage, and this is the stage where TIES is useful.
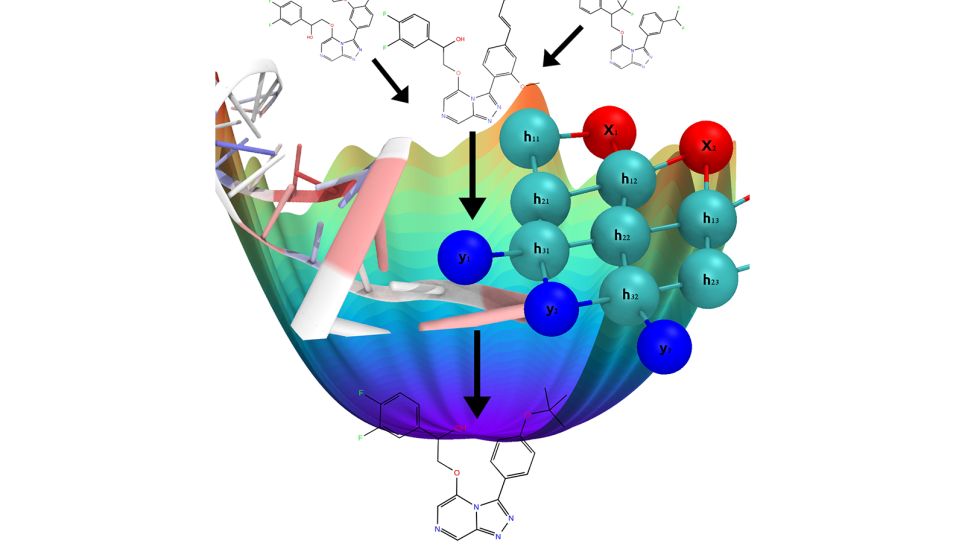
Approaching the problem from another direction, in the past two years new approaches to finding compounds useful for drug discovery have emerged using a form of AI known as machine learning. Researchers have applied machine learning to modeling compound-to-virus protein binding independently of physics-based simulations. These machine learning methods are much faster. “They are very, very quick. You can compute millions of compounds in a few hours,” Bhati says. But because these methods are dependent on training data in order to make useful predictions, the amount of data needed is huge.
In the case of drug discovery processes using unknown proteins, there is rarely enough data for them to be useful. This is a major impediment to using machine learning methods separately from physics-based simulations. Physics-based simulations do not depend upon input data, they are calculated based on theory and knowledge of the structure of the protein and the structure of the compound. When looking at millions or billions of compounds they become far too computationally expensive to be feasible. Machine learning methods require too much data to be useful on their own and physics-based simulations require too much computation to be useful on their own. “What people are thinking about now,” Bhati says, “is that each method overcomes the drawbacks of the other.”
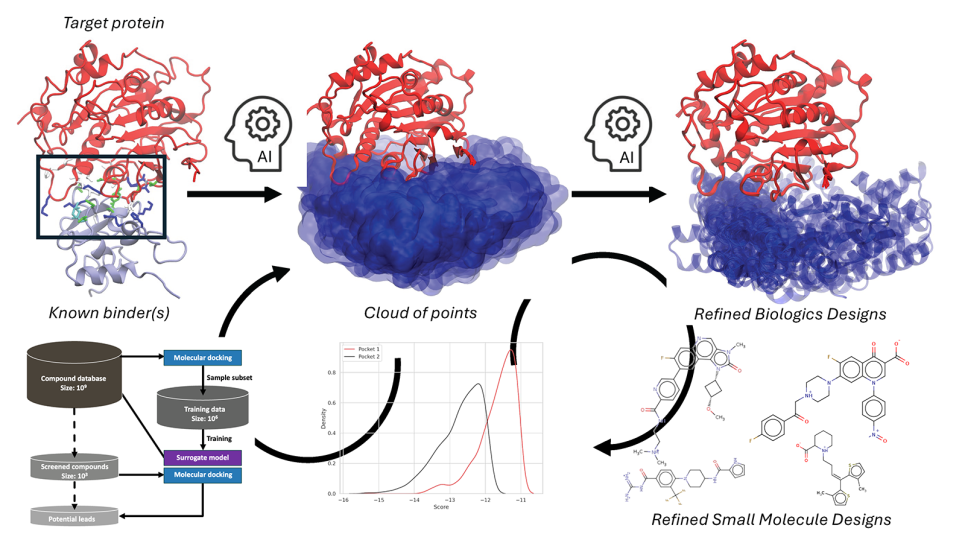
To avoid these problems, researchers are now combining the two methods. The researchers use physics-based simulations to determine the physical properties of compounds to use as training data for the machine learning algorithm, which then predicts potentially better molecules, which in turn are analyzed using physics-based simulations and then given back to the machine learning algorithm. This continues in a loop until it produces a short list of compounds likely to bind with the protein, at which point more computationally intensive physics-based simulations are applied.
This combined process requires a massive number of computations and needs powerful supercomputers in order to run. With an award from DOE’s Innovative and Novel Computational Impact on Theory and Experiment (INCITE) program, the team has access to the ALCF’s Theta system, and plans to leverage the upcoming Aurora exascale supercomputer when it becomes available. The INCITE award also pairs research teams with an in-house expert, in this case, Argonne computational scientist Wei Jiang who helped build and debug the code to enable the computations at the ALCF.
The ALCF machines are suited to this drug discovery pipeline, Bhati explains, because they “have specific hardware which is optimized for running machine learning algorithms in a nice way and also have high-performance computing infrastructure where we can run a large number of simulations in parallel.” Running simulations in parallel is important because, as Coveney says “the molecular dynamics is chaotic, every initial condition you use is different from each other one. And since the trajectories inside the simulation then diverge exponentially fast, you will always get different results from a single simulation to the next. You need to run ensembles of these things to get something statistically robust.”
As part of the INCITE project, the team collaborated with researchers at Argonne and across the country to develop a drug discovery pipeline called IMPECCABLE (Integrated Modeling PipelinE for Covid Cure by Assessing Better Leads) that is being used to look for drugs to treat COVID-19. IMPECCABLE is a modular workflow with different modules for physics-based and machine learning analyses that are connected together and loop in a specific pattern. IMPECCABLE combines both long-established physics-based simulations and new machine learning methods that are in their initial stages. “The overall workflow is comprised of about four separate workflows which are already large scale,” Coveney says.
These workflows cycle through simulations and machine learning methods. The workflow employs the TIES method to make physics-based results more accurate and reproducible. By the end of this process, Coveney says, “you will have what you think is your best ranking and that’s all done in silico and potentially within 24 hours, that’s quick—you could do experiments on them, to test them, getting it out of the in silico into the lab.”
This new, combined approach has substantially streamlined and sped up the process of drug discovery, allowing faster identification of compounds that bind to active sites on viruses.
The next steps for this pipeline include optimizing the newest modules and working on the workflow mechanisms between modules. IMPECCABLE has been tested on COVID targets, and once it is optimized, it will be used more regularly by the INCITE team and others to look for drugs for COVID and other viruses.
“The spirit of this,” Coveney says, “is to turn on its head the conventional way of doing pharmaceutical delivery, which is absolutely driven by experiments, typically making compounds, which is incredibly expensive. The reason for doing this is clearly to try and accelerate everything to reduce the amount of time you have to spend on experiments and focus only on compounds that look really potentially very good.”
==========
The Argonne Leadership Computing Facility provides supercomputing capabilities to the scientific and engineering community to advance fundamental discovery and understanding in a broad range of disciplines. Supported by the U.S. Department of Energy’s (DOE’s) Office of Science, Advanced Scientific Computing Research (ASCR) program, the ALCF is one of two DOE Leadership Computing Facilities in the nation dedicated to open science.
Argonne National Laboratory seeks solutions to pressing national problems in science and technology. The nation's first national laboratory, Argonne conducts leading-edge basic and applied scientific research in virtually every scientific discipline. Argonne researchers work closely with researchers from hundreds of companies, universities, and federal, state and municipal agencies to help them solve their specific problems, advance America's scientific leadership and prepare the nation for a better future. With employees from more than 60 nations, Argonne is managed by UChicago Argonne, LLC for the U.S. Department of Energy's Office of Science.
The U.S. Department of Energy's Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, visit https://energy.gov/science