Conducting Multiscale Modeling of Energy Storage Materials for Fuel Cells, Batteries
A growing consensus indicates that reliance upon fossil fuels not only threatens the climate patterns of the planet, but also the energy security of the United States. Given that the transportation sector accounts for a significant source of greenhouse gas emissions and is heavily influenced by foreign oil markets, the need to target the development of domestic, mobile clean energy technologies is evident. While several alternatives exist, advanced electrochemical systems, such as fuel cells and lithium-ion batteries, have been proposed for direct applications to automobiles. Some progress has been made in using battery packs to replace the internal combustion engine, as evidenced by the introduction of hybrid and all-electric vehicles such as the Chevy Volt.
In spite of these encouraging advances, electrochemical technologies continue to lag behind fossil fuels in performance and cost primarily as a result of the limitations imposed by the materials involved. Overcoming these limitations—the primary barrier to transformative breakthroughs in energy technology—requires the development of new materials.
Typically, designing new materials consists of inefficient “guess and check” approaches to optimize properties. Alternatively, computational modeling can be used to inform experimental efforts to minimize wasted resources and guide material design. The variety of important length and time scales associated with electrochemical phenomena conspires to make the modeling of electrochemical systems uniquely challenging, since the simulation scales cannot be feasibly addressed using one atomistic-scale methodology.
A team of scientists at The University of Chicago and Argonne National Laboratory, led by Gregory A. Voth, are working to overcome these challenges. The researchers are combining a powerful multiscale simulation methodology with the leadership-computing resources provided by the Argonne Leadership Computing Facility (ALCF) through the U.S. Department of Energy’s Innovative and Novel Computational Impact on Theory and Experiment (INCITE) program. The methodology being developed will address a variety of questions concerning the poorly understood ion-conduction mechanisms in both fuel cell membranes and at battery interfaces. It will form the first step in a potential feedback loop with experimental efforts.
Harnessing Expertise
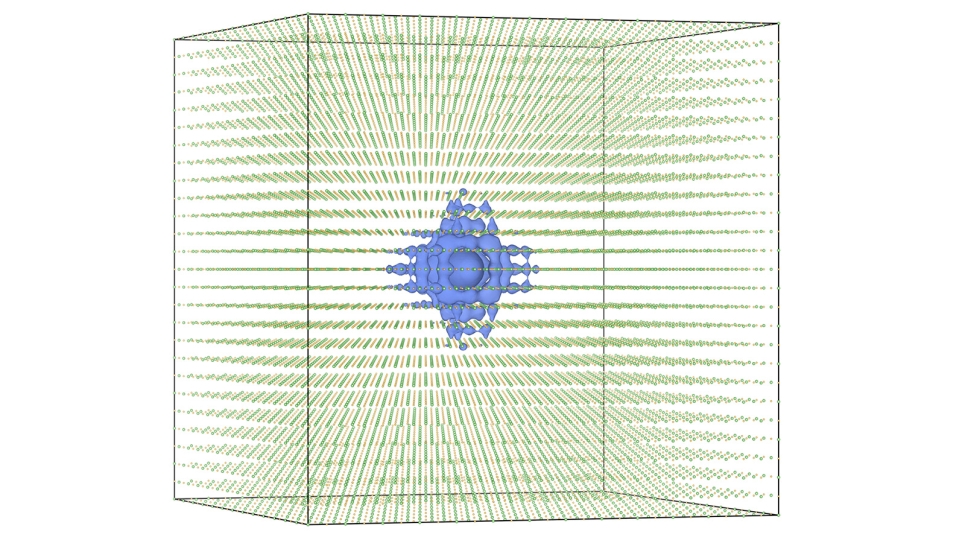
The basic concept of this multi-scale approach is to harness expertise at different levels of modeling, ranging from quantum mechanical methods that explicitly treat interactions with and between electrons of several molecules all the way up to modeling effective interactions between large collections of molecules found in proteins and materials. The connections between vastly different length scales are derived using a variational principle rooted in statistical mechanics, which allows the team to rigorously bridge detailed atomistic information to the properties of materials at length scales relevant to material design and fabrication. By combining accurate calculations on small systems that capture the essential physics and chemistry of the process under investigation, the multi-scale methodology can be applied to develop realistic representations of much larger systems (thousands of times larger), which can then themselves be used as the basis for developing even coarser models. Hence, each level of “resolution” can be connected to the next lowest or highest level in a consistent fashion.
The resulting coarse-grained models are significantly more computationally efficient than the models at higher resolutions and thus enable one to perform simulations at length and time scales far beyond what is originally possible at the highest resolution, but still retain information from quantum simulations. With the power of the ALCF's Blue Gene/P supercomputer used as the engine for these simulations, new system size and time scales can be reached, aiding in both the understanding of experimental results, as well as the validation and development of new theories.
Addressing Unique Challenges
Both fuel cell membranes and Li-ion battery interfaces carry unique challenges that require the development of multi-scale methods coupled to extensive computer resources. For fuel cell membranes, the mechanism for the transport of ions, which gives rise to the measured conductivity, involves detailed motions of the charge-carrying species (atomistic detail) within the overall structure of the full membrane, which creates intricate networks of water molecules that the ions must traverse. While atomistic simulations large enough to fully model the complex network of water “paths” for the ions are not realistic for sufficiently long times, results from simulations on representative systems (millions of atoms) can be used as input to the multi-scale methodology to develop coarse-grained models for the polymer membrane and water molecules. By exploring these materials at larger length scales and longer time scales than previously possible, the team can statistically converge physical properties and gain greater insight into how water and ions are transported in these complex materials.
For Li-ion batteries, the electrode-electrolyte interface is poorly understood as a result of a complex interplay between transport processes and numerous chemical reactions with the electrolyte components. In spite of the importance of these interfaces in battery operation, relatively little modeling has been conducted, and additional effort is required for the highest-resolution models. The research team has been developing molecular models for the solvent and reaction products at the interface to describe the important process of Li-ion migration from the electrolyte to the electrode surface.
An additional challenge is ensuring that the correct physics have been incorporated into the simulations. For this particular system, the effort warrants the inclusion of an electric field present during battery charging and discharging, which affects the structure at the electrode-electrolyte interface. The team has recently developed a method to include the effects of this electric field, and thus accurately model the electrode-electrolyte interface. Using the results from these atomistic simulations as input to the multi-scale methodology, in conjunction with the leadership computing resources, the study of Li-ion dynamics will be made possible at length scales previously unexplored.
Advancing the Research
After the team of researchers has completed the foundational atomistic simulations for the fuel cell membranes, coarse-grained models will be constructed and used to accelerate simulations of same-sized systems as well as begin initial studies on membrane structure at much larger length-scales. These initial atomistic simulations are also currently being analyzed to examine the degree by which water content alters the solvation structure of charged defects and ionic sidechains along the polymer backbone.
For the Li-ion battery systems, the team has parameterized Li-ion interactions with electrolytes and salts using electronic structure calculations and the models are currently being validated. Results from these initial studies already highlight the importance of including the correct physics for describing the electric field at the electrolyte/electrode interface with observed changes in Li-ion and electrolyte solvation structures near the interface under different applied voltages. Extending the accessible time and length scales for simulating these systems will enable the exploration of fundamental questions regarding proton, hydroxide, and Li-ion transport.
Domains
Allocations