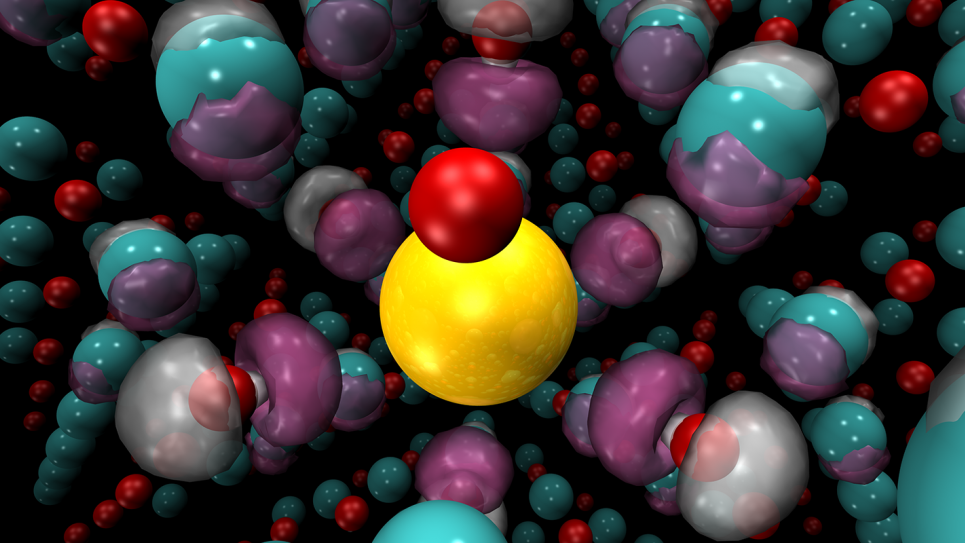
Recent advances in quantum Monte Carlo (QMC) methods have the potential to revolutionize computational materials science, a discipline traditionally driven by density functional theory (DFT). While DFT—an approach that uses quantum-mechanical modeling to examine the electronic structure of complex systems—provides convenience to its practitioners and has unquestionably yielded a great many successes throughout the decades since its formulation, it is not without shortcomings, which have placed a ceiling on the possibilities of materials discovery. QMC is poised to break this ceiling.
The key challenge is to solve the quantum many-body problem accurately and reliably enough for a given material. QMC solves these problems via stochastic sampling—that is, by using random numbers to sample all possible solutions. The use of stochastic methods allows the full many-body problem to be treated while circumventing large approximations. Compared to traditional methods, they offer extraordinary potential accuracy, strong suitability for high-performance computing, and—with few known sources of systematic error—transparency. For example, QMC satisfies a mathematical principle that allows it to set a bound for a given system’s ground state energy (the lowest-energy, most stable state).
QMC’s accurate treatment of quantum mechanics is very computationally demanding, necessitating the use of leadership-class computational resources and thus limiting its application. Access to the computing systems at the Argonne Leadership Computing Facility (ALCF) and the Oak Ridge Leadership Computing Facility (OLCF)—U.S. Department of Energy (DOE) Office of Science User Facilities—has enabled a team of researchers led by Paul Kent of Oak Ridge National Laboratory (ORNL) to meet the steep demands posed by QMC. Supported by DOE’s Innovative and Novel Computational Impact on Theory and Experiment (INCITE) program, the team’s goal is to simulate promising materials that elude DFT’s investigative and predictive powers.
To conduct their work, the researchers employ QMCPACK, an open-source QMC code developed by the team. It is written specifically for high-performance computers and runs on all the DOE machines. It has been run at the ALCF since 2011.
Functional materials
The team’s efforts are focused on studies of materials combining transition metal elements with oxygen. Many of these transition metal oxides are functional materials that have striking and useful properties. Small perturbations in the make-up or structure of these materials can cause them to switch from metallic to insulating, and greatly change their magnetic properties and ability to host and transport other atoms. Such attributes make the materials useful for technological applications while posing fundamental scientific questions about how these properties arise.
The computational challenge has been to simulate the materials with sufficient accuracy: the materials’ properties are sensitive to small changes due to complex quantum mechanical interactions, which make them very difficult to model.
The computational performance and large memory of the ALCF’s Theta system have been particularly helpful to the team. Theta’s storage capacity has enabled studies of material changes caused by small perturbations such as additional elements or vacancies. Over three years the team developed a new technique to more efficiently store the quantum mechanical wavefunctions used by QMC, greatly increasing the range of materials that could be run on Theta.
Experimental Validation
Kent noted that experimental validation is a key component of the INCITE project. “The team is leveraging facilities located at Argonne and Oak Ridge National Laboratories to grow high-quality thin films of transition-metal oxides,” he said, including vanadium oxide (VO2) and variants of nickel oxide (NiO) that have been modified with other compounds.
For VO2, the team combined atomic force microscopy, Kelvin probe force microscopy, and time-of-flight secondary ion mass spectroscopy on VO2 grown at ORNL’s Center for Nanophase Materials Science (CNMS) to demonstrate how oxygen vacancies suppress the transition from metallic to insulating VO2. A combination of QMC, dynamical mean field theory, and DFT modeling was deployed to identify the mechanism by which this phenomenon occurs: oxygen vacancies leave positively charged holes that are localized around the vacancy site and end up distorting the structure of certain vanadium orbitals.
For NiO, the challenge was to understand how a small quantity of dopant atoms, in this case potassium, modifies the structure and optical properties. Molecular beam epitaxy at Argonne’s Materials Science Division was used to create high quality films that were then probed via techniques such as x-ray scattering and x-ray absorption spectroscopy at Argonne’s Advanced Photon Source (APS) for direct comparison with computational results. These experimental results were subsequently compared against computational models employing QMC and DFT. The APS and CNMS are DOE Office of Science User Facilities.
So far the team has been able to compute, understand, and experimentally validate how the band gap of materials containing a single transition metal element varies with composition. Band gaps determine a material’s usefulness as a semiconductor—a substance that can alternately conduct or cease the flow of electricity (which is important for building electronic sensors or devices). The next steps of the study will be to tackle more complex materials, with additional elements and more subtle magnetic properties. While more challenging, these materials could lead to greater discoveries.
New chemistry applications
Many of the features that make QMC attractive for materials also make it attractive for chemistry applications. An outside colleague—quantum chemist Kieron Burke of the University of California, Irvine—provided the impetus for a paper published in Journal of Chemical Theory and Computation. Burke approached the team’s collaborators with a problem he had encountered while trying to formulate a new method for DFT. Moving forward with his attempt required benchmarks against which to test his method’s accuracy. As QMC was the only means by which sufficiently precise benchmarks could be obtained, the team produced a series of calculations for him.
The reputed gold standard for many-body system numerical techniques in quantum chemistry is known as coupled cluster theory. While it is extremely accurate for many molecules, some are so strongly correlated quantum-mechanically that they can be thought of as existing in a superposition of quantum states. The conventional coupled cluster method cannot handle something so complicated. Co-principal investigator Anouar Benali, a computational scientist at the ALCF and Argonne’s Computational Sciences Division, spent some three years collaborating on efforts to expand QMC’s capability so as to include both low-cost and highly efficient support for these states that will in future also be needed for materials problems. Performing analysis on the system for which Burke needed benchmarks required this superposition support; he verified the results of his newly developed DFT approach against the calculations generated with Benali’s QMC expansion. They were in close agreement with each other, but not with the results conventional coupled cluster had generated—which, for one particular compound, contained significant errors.
“This collaboration and its results have therefore identified a potential new area of research for the team and QMC,” Kent said. “That is, tackling challenging quantum chemical problems.”
The research was supported by DOE’s Office of Science. ALCF and OLCF computing time and resources were allocated through the INCITE program.
Argonne National Laboratory seeks solutions to pressing national problems in science and technology. The nation's first national laboratory, Argonne conducts leading-edge basic and applied scientific research in virtually every scientific discipline. Argonne researchers work closely with researchers from hundreds of companies, universities, and federal, state and municipal agencies to help them solve their specific problems, advance America's scientific leadership and prepare the nation for a better future. With employees from more than 60 nations, Argonne is managed by UChicago Argonne, LLC for the U.S. Department of Energy's Office of Science.
The U.S. Department of Energy's Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, visit https://energy.gov/science