Theta provides insights into the building blocks of life
As part of the ALCF's Theta Early Science Program, researchers from the University of Chicago used the new supercomputer to advance the study of membrane transport proteins. Their work included an investigation of how pH conditions affect the passage of small drug molecules across cell membranes, lending insight into the origin of acidity/alkalinity effects on drug adsorption.
It is estimated that there are some 20,000 protein-coding genes in the human genome, each composed of a multitude of amino acids. These proteins are literally the building blocks of life, responsible for a myriad of functions. But few have the power of membrane transport proteins.
These biological “molecular machines” hold considerable interest for Benoît Roux, Amgen Professor, Department of Biochemistry and Molecular Biology, the University of Chicago.
“The proteins that are associated with biological membranes are particularly interesting because some of them can really function like little molecular machines, like little pumps,” exclaims Roux.
Membrane transport proteins are a unique class of large-molecule biological systems that play a major role in the dynamics of cell function. They help facilitate and control the passage of essential nutrients in or out of the cell to maintain its integrity. But the processes by which membrane proteins accomplish these feats are little understood.
Roux and a team of researchers from the University of Chicago and the University of Illinois at Urbana-Champaign are trying to paint an atomistic portrait of one family of large-membrane protein molecules called ion pumps. Through high-performance computer simulations, they are working to derive quantitative predictions of the energies these pumps produce and consume. Understanding their dynamics could lead to the design of drugs that affect specific functions.
The project is part of the Argonne Leadership Computing Facility’s (ALCF) Theta Early Science Program (ESP), which allowed a select group of researchers to work on Theta, the facility’s 11.69-petaflops Intel-Cray supercomputer, before it was released to the larger research community last July. The ALCF is a DOE Office of Science User Facility.
Roux’s team continues to use Theta to build a scalable computational strategy that combines classic and novel molecular dynamics (MD) algorithms.
Investigating the impact of pH
The function of these large membrane protein molecules results from molecular processes associated with conformational transitions that occur in or because of the surrounding chemical environment.
For example, pH, the acidity/alkalinity scale, plays an important role in cellular environments. Nearly every biological process occurs in an aqueous environment and is therefore greatly influenced by the pH of their water environment. This includes the movement of drug molecules across cell membranes.
Researchers are taking a deeper look at the relationship between constant pH conditions and conformational transitions to better understand how the proteins carry out their function.
But in biology, experimental studies of conformational transitions associated with protein function are often difficult, as they are fast moving, short lived, and hard to measure. In addition, pH-dependent simulation studies present their own challenges given the large number of protonation sites, those that accept/donate hydrogen ions under various pH conditions. Each site on a macromolecule must be thoroughly sampled, necessitating scalable algorithms and their implementation in software.
The specific family of systems on which Roux’s team has been working are called P-type ATPases. These large membrane proteins bind the molecule adenosine triphosphate (ATP) and then spontaneously follow the breaking of the bond to harness a kind of chemical energy that is exploited to pump ions across the membrane.
“They are literally pumping ions,” explains Roux, “in the sense that they transport ions against their electrochemical gradient. That means you’re taking ions from places where there are not a lot of them and you’re pumping them into places that have a lot of them. So you’re doing something that is truly work, something against the simple thermodynamic equilibrium.”
The specific pump that Roux and his cohorts are looking at is called the sodium/potassium, or Na/K, pump.
All of the living cells in our body are abundant in potassium, but not sodium, which is ample in blood plasma. As elucidated by Nobel Prize-winning chemist Walther Hermann Nernst over a century ago, this imbalance of ion concentrations creates an electrostatic potential across the membrane and is critical for everything in living systems from propagation of the nerve impulse and muscle contraction, to secretion, development of the embryo, and much more.
Almost half of all ATP consumed by the human body is used by the Na/K pump.
Determining how the P-type ATPases works helps researchers understand how to keep these functions in balance and develop corresponding treatments and medications. The widely used anti-acid medication Omeprazole, for example, is a drug that reduces acidity in the stomach by inhibiting a P-type proton pump that works very much like the Na/K pump.
Simulating atomic pathways
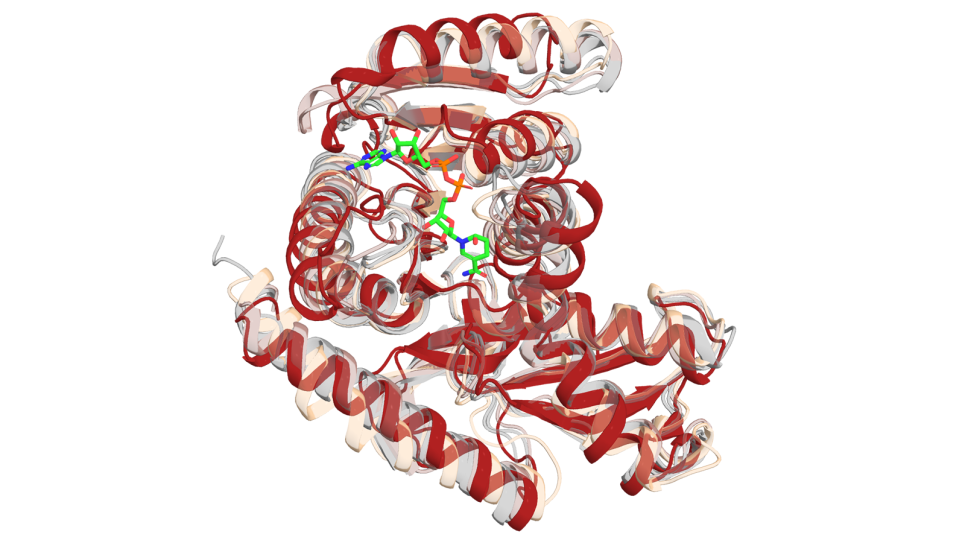
The difficulty lies in identifying the different 3D atomic structures associated with each protein, which have many states, where different elements of the protein are organized. Biochemists determine these structures through X-ray crystallography. In the case of the Na/K pump, a handful of X-ray structures are currently known.
To elucidate the atomic pathways in between the known X-ray structures, the team uses a modified version of the nanoscale molecular dynamics (NAMD) code, designed for high-performance simulations of large biomolecular systems.
“It’s a little bit like going on a trek through the Himalayas. The crystals represent the structures where you know you’re going to sleep at night—these are our milestones. But you don’t really know what’s happening between each structure,” says Roux.
Simulations will build upon existing structural data derived from crystallography. Missing data about the conformational pathways between the known structures will be predicted by a simulation technique called string method, which takes us back to our cabins in the Himalayas.
Akin to a chain of Sherpas connected by ropes between each of those cabins, string method takes the path of least resistance—seeking to go through the most favorable mountain pass—thus describing the most plausible pathways from one shelter to another.
“So we use the string method to understand how the different structures are related to one another. They map out how you can go from one conformational state to the other,” says Roux.
ALCF staff has been working to optimize the newer code to run seamlessly on Theta, and develop the overall computational methodology for the project. This will include more novel approaches, such as the aforementioned string method and hybrid MD/Monte Carlo simulations. The implementation is massively parallel and works efficiently for large biomolecules at realistic pH conditions.
“Using Theta has allowed us to not only prototype and test new and complicated algorithms that have never been implemented before, but also immediately apply those methods to exciting and challenging problems that would not have been available on smaller and less sophisticated computers,” says ALCF postdoctoral appointee Brian Radak, an ESP project member.
Preliminary simulations reveal how pH conditions affect the passage of small drug molecules across cell membranes, lending insight into the origin of acidity/alkalinity effects on drug adsorption. The scalable and generalizable feature of the constant-pH method opens the door to pH-dependent drug optimization and discovery.
Computing time and resources for this work were allocated through the ALCF’s Theta Early Science Program. The ALCF is supported by the DOE Office of Science’s Advanced Scientific Computing Research program.